- 移动端


上海艾跃(Active Motif)生物科技有限公司品牌商
10 年
手机商铺
- NaN
- 0.2999999999999998
- 0.2999999999999998
- 2.3
- 2.3
上海艾跃(Active Motif)生物科技有限公司
入驻年限:10 年
- 联系人:
Active Motif
- 所在地区:
上海 闵行区
- 业务范围:
试剂、实验室仪器 / 设备、技术服务、抗体
- 经营模式:
生产厂商 经销商
推荐产品



公司新闻/正文
【新品上市】RNA-Seq文库制备试剂盒!快速!简单!低成本!
1302 人阅读发布时间:2022-07-21 13:59
在过去的十年里,RNA-seq已然成为研究转录的基础技术,解析高分辨率的转录信息。
近几年随着单细胞技术的诞生,使得现RNA-seq的分辨率进一步提高,能够获得单个细胞的转录信息。
在疾病和癌症研究中,转录组数据可以深入了解从转录调控和细胞分化到致病或致癌的整个转录过程。
而为获得成功的RNA-Seq数据分析,RNA的高质量文库制备是必不可少的。
虽然现在市面上已经开发出各种用于文库构建的方法步骤,但它通常都涉及RNA片段化、将RNA转化为互补DNA(cDNA)、添加adapter以及将文库扩增到足以进行NGS测序的数量。
虽已有一些减少文库准备步骤所需时间和成本的方法相继问世,但其转录本的测序深度要求依旧保持着不变,通常为20-30M或更多的双端reads (PE reads)。
虽然测序成本在不断下降,但它仍然相当昂贵,尤其是面对数百个样本时,这将是一笔不菲的费用。
有一种鲜为人知的文库制备替代方法,它可以在不牺牲高质量基因表达谱数据的情况下大大降低测序深度要求。
这种方法被称为3’-Digital Gene Expression(3’-DGE),只需要3-10 M的单端 Reads,虽然它不适用于所有RNA-seq应用,但如果使用得当,其不失为一种以更低成本进行差异表达分析和定量转录组分析的优良方法。
什么是3’-Digital Gene Expression (3’-DGE) RNA-seq ?
高通量Bulk RNA-Seq有许多应用,包括转录组组装(Transcriptome assembly)、可变剪切(Alternative splice)或转录亚型 (Transcript isoforms)、转录变体(Transcript variant)的检测和分析等位基因特异性表达 (Allele-specific expression)。
然而,典型RNA-Seq项目的主要目标是识别在某些实验条件下上调或下调的基因和通路。
虽然有许多技术用于RNA-Seq实验,但3’-Digital Gene Expression(3’-DGE)是一种相对新的RNA-Seq模式,以每个转录本的3’-末端为目标,并对差异基因表达进行优化。
Digital Gene Expression中的 “digital” 是指counting,这种RNA测序方法有时被称为“转录计数(Transcript counting)”法。
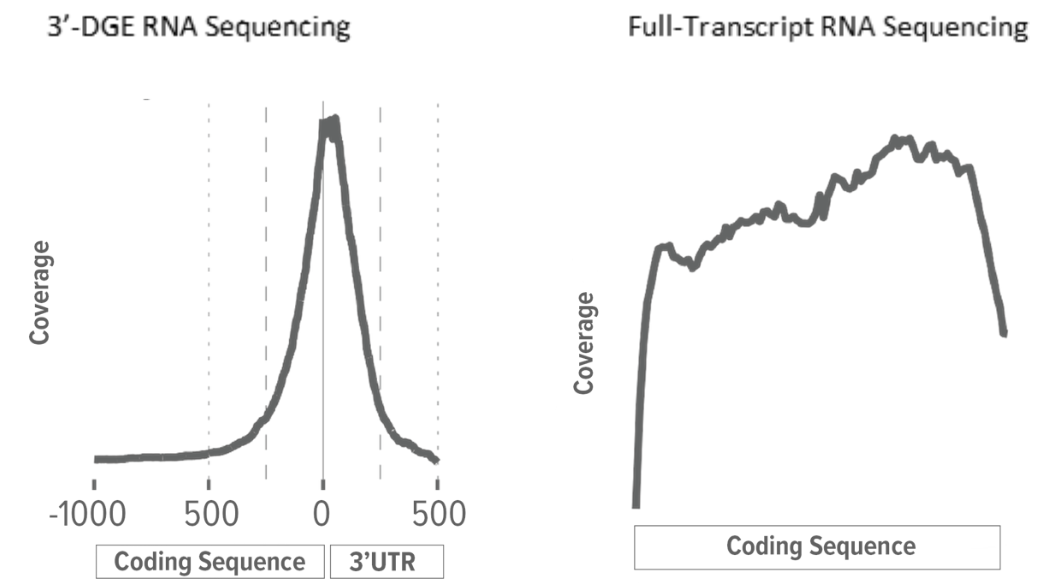
3’-DGE与全转录RNA测序相比如何?
传统的全转录(FT)覆盖RNA-Seq产生的测序reads能够完整映射到整个表达的转录组,这是通过RNA随机片段化和cDNA合成步骤使用随机引物实现的。
当映射到参考转录组或基因组时,测序获得的reads能够在外显子之间均一分布。
对于传统的RNA-Seq,映射到基因的Reads是转录丰度和转录长度的函数。
因此,与转录物丰度相似的短基因相比,长基因将有更多的Reads映射到它。
与随机合成相比,3’-DGE RNA-Seq的cDNA合成是以带有oligo (dT) 的adaptor为起始与转录本的poly(A)尾部对应。
这意味着3’-DGE输出的reads往往只映射到转录本的3’-末端。
由于转录分子和cDNA分子之间存在理论上的1:1相关性,转录长度无关紧要,映射到基因的Reads数仅是转录丰度的函数。
因此,一个长基因与一个转录物丰度相似的短基因具有相当的Reads数。
3’-DGE RNA-Seq的使用有三个主要的限制因素。
首先,3’-DGE仅限于真核生物,因为第一条链的合成依赖于真核mRNA链的ploy (A) 尾。
由于大多数3’-DGE的Reads需要映射到基因的3’-UTR部分,因此比对的基因组需要比较完善的注释。
但对于具有已知翻译终止位点但没有注释转录终止位点的物种,可以基于翻译终止位点上游和下游的距离,以生物信息学的方式设置具有感兴趣区域的参考序列或注释。
另一方面,为了避免错估3’-UTR长度相关的问题,最变通的解决方法是采用链特异性RNA测序,以区分映射反义链上相邻基因附近3’-UTR的Reads。
第三个限制与分析灵活性有关,进行分析需要来自整个转录本(组装、剪接等)的信息的研究人员可能更倾向于使用传统的RNA-Seq或使用这两种方法的战略性组合。
考虑到这些局限性,或由于研究人员对3’-DGE的优势不够了解,所以可能更多的研究人员目前不使用3’-DGE作为他们RNA-Seq的优先选择。
由于3’-DGE所产生的cDNA与转录本是一对一的关系,使得3’-DGE的转录本计数方法更加高效,减小了分析的复杂性。
例如,如果我们获得了100 个reads的RNA-Seq结果都映射到了基因A上。
若是传统的RNA-Seq,我们并不能知道准确知道是多少A基因的转录量(是1?、10?、25?、或是更多?)产生了这100个reads,即使做了转录本长度归一化(Normalization),这种不确定性也是真实存在的。
然而,在3’-DGE实验中,这100个Reads基本上就对应于100个转录拷贝量。
3’-DGE在转录本统计这一方面降低了分析的复杂性,这可能是这种方法的最大优势。
转录本计数意味着与传统RNA-Seq相比,获得优良数据分析所需要的测序深度更低。
此外,双端测序并不会使得3’-DGE获得更多有价值的信息。
因此,每个样本3-10 M的单端Reads的3’-DGE测序足以用于下游分析(传统RNA-Seq可能需要20-30 M reads的双端测序量),可以显著降低测序预算。
发掘3’-DGE的更多可能
潜在的核糖核酸酶会对RNA提取产生不可控的影响,传统的RNA-Seq需要高质量的RNA样本,通常QC结果的RIN(RNA完整性数)需要大于7才能满足RNA-seq的需求。
对于一些稀缺样本或特殊的样本,想使RIN>7往往很难达到,虽然这可能会对依赖完整转录本的传统RNA-Seq建库造成阻碍,但3’-DGE文库对这种变异性不太敏感。
无论采用何种RNA提取方法,都不可能保证从某些样本类型中获得高质量的RNA,在这种情况下,3’-DGE有可能比常规RNA-Seq更易获得良好准确的差异基因表达结果。
变异检测有可能为全面的RNA-Seq分析增加新的维度。
鉴于编码蛋白的外显子区域比基因的非编码区承受更大的选择压力,3’-DGE获得的Reads与传统RNA-Seq相比更易富集变异。
这些额外的变异信息可用于基因表达研究以外的分析,包括表征BIL/NIL(回交自交系/近等基因系)和进行GWAS或QTL定位。
最近的一项研究利用了这种降低的选择压力,并使用3’-UTR最终解析了鸟类的进化树。
3'-DGE这种方法具有许多优点,包括测序成本大幅降低、更容易的数据分析以及对RNA质量的敏感性较低。
所以,使用我们Active Motif的YourSeq (FT&3'DGE) Strand-Specific mRNA建库试剂盒开始您的3'-DGE测序吧:
-
YourSeq (FT&3'DGE) Strand-Specific mRNA建库试剂盒
-快速、灵活的建库试剂盒
-用于全长和3’ Digital Gene Expression RNA测序
详情请扫描下图中二维码:

https://www.activemotif.com.cn/catalog/1358/yourseq-mrna-library-prep
3’-Digital Gene Expression(3’-DGE)测序
轻松get!
如果您有任何疑问,
欢迎联系021-20926090,
18521362870。